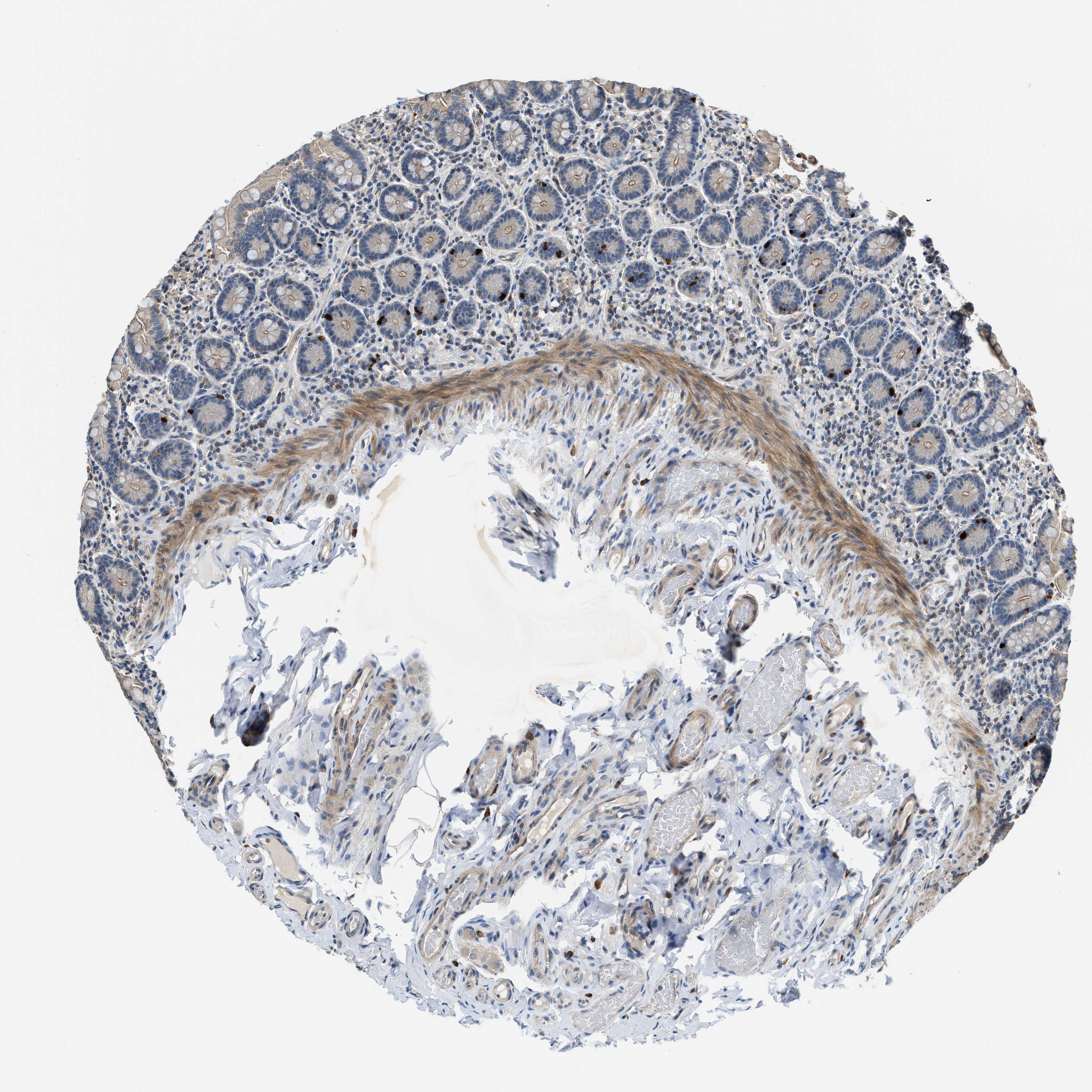
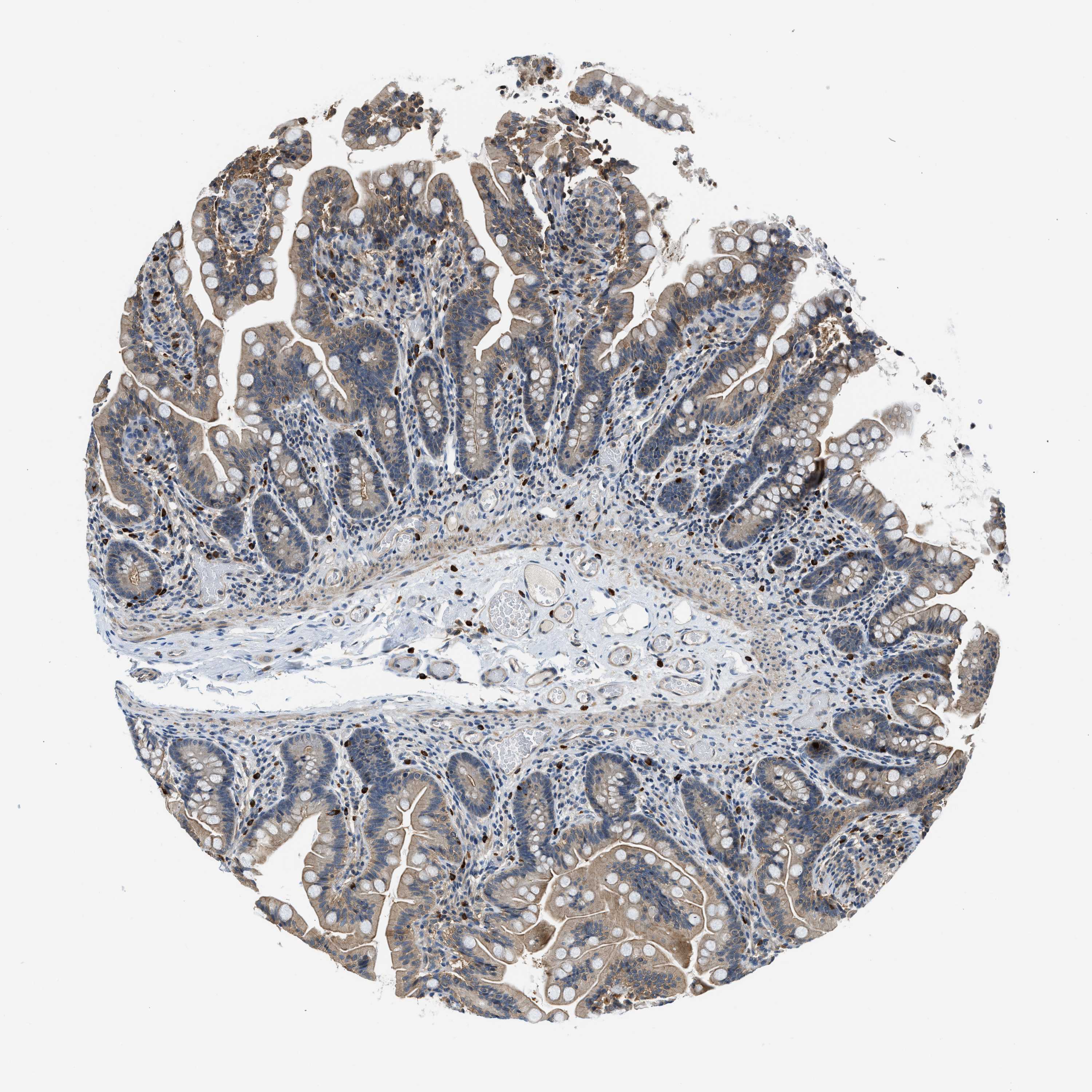
The detailed normal tissue page shows images of the stained tissue, together with antibody staining/expression level of the cell types.
If a
knowledge-based annotated protein expression is provided, this is reported for each cell
type at the top of the page and the staining level of the individual antibodies is given underneath each antibody ID.
Samples from up to three different individuals have been stained for each antibody. The gender, age and
tissue characterization
are reported for each individual (patient) and are viewable when clicking on the image for magnification.
The images can be clicked for an enlarged view that can be panned. From the enlarged view, all stained images for all
antibodies can be browsed (represented by miniature images). The miniature image with an orange overlay is the currently displayed image.
For histological reference, view
small intestine in the
histological dictionary.
















 Male, age 65
Male, age 65

 Male, age 39
Male, age 39
 Male, age 68
Male, age 68
 Male, age 85
Male, age 85